
We are a multidisciplinary team bringing together expertise in chemistry, physics, materials science, computer science, and engineering. Explore our work and join us!

Methods development
Our research group specializes in the development and application of novel molecular simulation methods to investigate activated processes, with a particular focus on complex catalytic reactions.
More precisely, our work focuses on a variety of free energy methods and on the design of descriptors, such as collective variables, to describe intricate chemical reactions occurring in complex environments. To tackle these challenges, we combine modern data-driven approaches and machine learning techniques with state-of-the-art molecular simulation methods. We are also actively engaged in the field of automated exploration of chemical reactivity, employing molecular dynamics-based approaches to uncover new reaction pathways without prior mechanistic assumptions. To access long time scales and ensure exhaustive sampling, we develop and train in-house machine learning interatomic potentials, grounded in ab initio data. These potentials are constructed using advanced active learning schemes, allowing us to reach high levels of accuracy while maintaining computational efficiency.
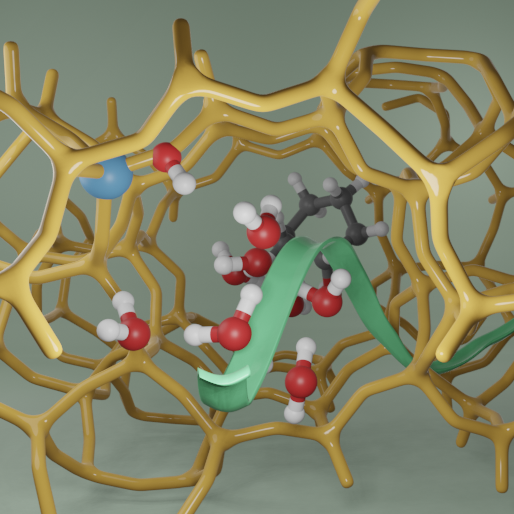
Heterogeneous catalysis
Heterogeneous catalysis is a cornerstone of the modern chemical industry, enabling the large-scale production of fuels, chemicals, and materials essential to everyday life. However, a detailed molecular-level understanding of how catalysts function—particularly under realistic (operando) conditions involving temperature, pressure, and complex reaction environments—remains a significant challenge. Our group is dedicated to providing this molecular insight by combining advanced molecular simulations with data-driven analysis. We employ free energy methods and machine learning interatomic potentials, rigorously trained on high-level ab initio data, to simulate catalytic processes with high accuracy and computational efficiency. These simulations allow us to access both thermodynamics and kinetics, shedding light on the conditions that govern catalytic activity and selectivity. To extract detailed mechanistic information, we integrate automated data analysis workflows and reaction discovery tools, which enable us to map out reaction networks, identify rate-limiting steps, and reveal hidden pathways that are not always accessible experimentally.Through this approach, we aim to contribute to the rational design and optimization of catalytic materials, providing fundamental understanding that can guide the development of more efficient, selective, and sustainable catalytic processes.
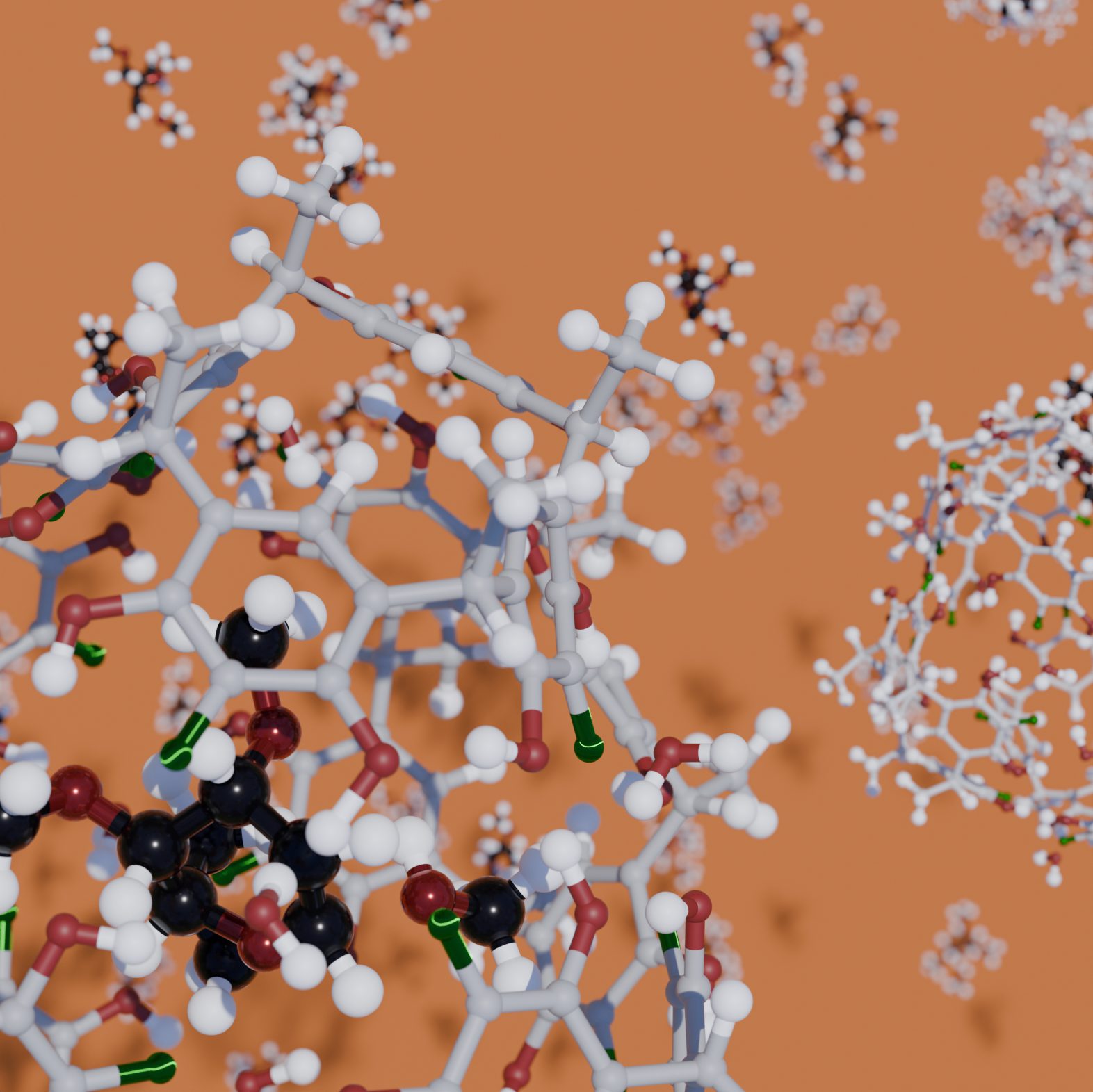
Supramolecular Catalysis
Supramolecular systems represent a powerful and elegant platform for catalysis, often emulating the remarkable efficiency and selectivity of natural enzymes. Their ability to create well-defined molecular environments—through non-covalent interactions such as hydrogen bonding, π-stacking, and host–guest recognition—makes them ideal candidates for controlling reactivity in confined spaces. However, their structural complexity, flexibility, and dynamic behavior make them exceptionally challenging to model and understand at the molecular level. Our group addresses these challenges through a multiscale modeling strategy that combines quantum chemical methods, free energy calculations, and post-transition state dynamics. This integrative approach enables us to capture both the static and dynamic aspects of supramolecular systems, revealing how subtle structural features influence reactivity, selectivity, and catalytic efficiency. By leveraging machine learning tools and advanced sampling techniques, we can efficiently explore complex energy landscapes and uncover reaction pathways that are otherwise difficult to predict. Our simulations allow us to estimate activation free energies, binding affinities, and reaction mechanisms with high fidelity.